1. Data.
The data I used was 53 fastq files taking up about 300G of space. I didn't plan on doing anything with the raw data.
2. Mapping:
I used my branch of blasr (https://github.com/mchaisso/blasr/commit/c950018b65728cf9a0806381f1e69fba6ea40b08)
to map to build 38, using a precomputed index
blasr input.fofn GRCh38.fasta -maxMatch 25 -advanceExactMatches 5 -sam -nproc 16 -minMapQV 30 -out /dev/stdout | samtools view -bS - | samtools sort -T /tmp/clive -o clive.bam
This took < 1 day using 16 cores and about 30G of memory.
3. Basic sequencing stats. The dataset had about 30X euchromatic coverage, with about 0.5Gbase unmapped. The data release quotes 50X, so there is a chance I didn't grab all the files.

5. Calling structural variation.
The current pipeline I use for calling SV on a diploid sample is: 1. phase SNVs. 2. Partition SMS reads counting overlap of phased SNVs, and 3. Calling SV on each set of partitioned reads separately. I didn't try taking the time to do the phasing because the SNV strand bias would cause a bunch of artifacts to appear in phase. The code to separate strand bias from real SNVs represented on both strands is running now, but it's /very/ slow and probably won't finish until long past my attention has turned back to my normal research.
I modified my pipeline to do local assembly in 60kbp tiling windows using Canu V1.4. Instead of mapping the local assemblies, the 10 longest p-reads from each local assembly were mapped, this helps fix the problem of detecting heterozygous variants. The p-reads had an average accurcy of 96.9%, making them unsuitable for indel calling and OK for calling SV. The p-reads were mapped back to hg38, and a set of unmerged SVs were output simply by parsing the cigars strings. To merge, SVs were split according to insertion and deletion, and then a graph was made with each SV as a node, and an edge connecting any two SVs if they had 50% reciprocal overlap. The largest SV from any connected component with at least two nodes was output.
3. Basic sequencing stats. The dataset had about 30X euchromatic coverage, with about 0.5Gbase unmapped. The data release quotes 50X, so there is a chance I didn't grab all the files.

While there are a few very long (> 100kbp) sequences here, it only accounts for 0.2% of the total coverage, and 8% of the total coverage allowing reads over 50kbp. This implies we can't start assuming de novo assembly with 100kbp reads yet.
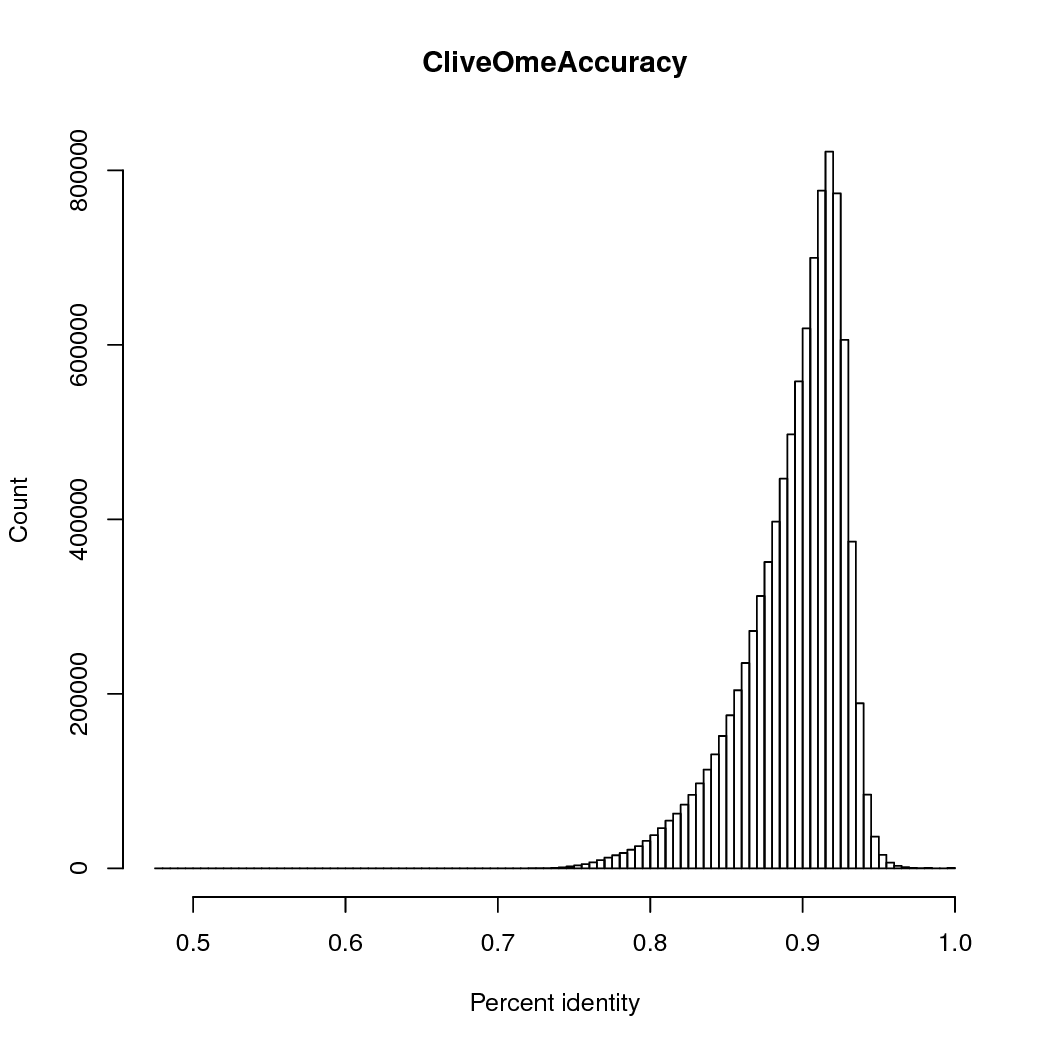
 The accuracy was 89%, counting # matches/ (#matches + # insertions + # deletions + # mismatches), so multi-base insertions or deletions are counted as single events.
The accuracy was 89%, counting # matches/ (#matches + # insertions + # deletions + # mismatches), so multi-base insertions or deletions are counted as single events.
 And like all SMS, there isn't a length/accuracy bias.
And like all SMS, there isn't a length/accuracy bias.
4. Sequencing error modes.
I'm a noob on nanopore sequencing, so much of what I may mention about error modes is probably well known. Generic regions look fine, but have a high SNV rate.

Zooming in on a few of these there was a pretty high strand bias for SNVs. Another way to look at this is, for each SNV, look at the ratio of the min(# forward, #reverse)/(#forward+#reverse) for all reads supporting an SNV, for the two haplotypes.

Clive mentioned on Twitter this could be due to modified bases and their basecaller not supporting native DNA yet, so I won't look any more to figure out what's happening.
Here is the strand ratio for the pacbio SNVs on chromosome 1 for a different individual:


4. Sequencing error modes.
I'm a noob on nanopore sequencing, so much of what I may mention about error modes is probably well known. Generic regions look fine, but have a high SNV rate.

Zooming in on a few of these there was a pretty high strand bias for SNVs. Another way to look at this is, for each SNV, look at the ratio of the min(# forward, #reverse)/(#forward+#reverse) for all reads supporting an SNV, for the two haplotypes.

Here is the strand ratio for the pacbio SNVs on chromosome 1 for a different individual:

Also, effectively all poly-A/polyT sequences I saw were deleted:


5. Calling structural variation.
The current pipeline I use for calling SV on a diploid sample is: 1. phase SNVs. 2. Partition SMS reads counting overlap of phased SNVs, and 3. Calling SV on each set of partitioned reads separately. I didn't try taking the time to do the phasing because the SNV strand bias would cause a bunch of artifacts to appear in phase. The code to separate strand bias from real SNVs represented on both strands is running now, but it's /very/ slow and probably won't finish until long past my attention has turned back to my normal research.
I modified my pipeline to do local assembly in 60kbp tiling windows using Canu V1.4. Instead of mapping the local assemblies, the 10 longest p-reads from each local assembly were mapped, this helps fix the problem of detecting heterozygous variants. The p-reads had an average accurcy of 96.9%, making them unsuitable for indel calling and OK for calling SV. The p-reads were mapped back to hg38, and a set of unmerged SVs were output simply by parsing the cigars strings. To merge, SVs were split according to insertion and deletion, and then a graph was made with each SV as a node, and an edge connecting any two SVs if they had 50% reciprocal overlap. The largest SV from any connected component with at least two nodes was output.
| Insertion | Deletion | |||||||
| Repeat type | Count | Mean | s.d. | Total | Count | Mean | s.d. | Total |
| Complex | 4968 | 118.91 | 191.23 | 590753 | 18616 | 83.75 | 121.19 | 1559030 |
| AluY | 1319 | 263.54 | 47.94 | 347611 | 1857 | 220.39 | 119.8 | 409259 |
| AluS | 183 | 176.63 | 79.68 | 32323 | 1589 | 95.21 | 57.34 | 151293 |
| L1Hs | 171 | 1971.84 | 2145.28 | 337185 | 259 | 1597.51 | 2240.74 | 413754 |
| L1 | 192 | 765.58 | 1695.6 | 146992 | 1734 | 148.16 | 393.03 | 256910 |
| L1P | 427 | 553.71 | 931.83 | 236436 | 2109 | 172 | 381.9 | 362747 |
| SVA | 256 | 709.81 | 841.54 | 181711 | 430 | 452.19 | 798.13 | 194442 |
| HERV | 44 | 288.02 | 422.82 | 12673 | 274 | 143.41 | 355.73 | 39295 |
| Alu-mosaic | 254 | 358.31 | 256.56 | 91010 | 589 | 166.86 | 195.64 | 98278 |
| STR | 4690 | 244.82 | 328.21 | 1148188 | 13312 | 84.65 | 65.21 | 1126812 |
| Satellite | 2583 | 297.44 | 482.81 | 768280 | 2645 | 221.41 | 460.67 | 585625 |
| Singletons | 308 | 208.39 | 367.86 | 64184 | 1898 | 103.76 | 140.33 | 196930 |
| Tandem_Repeats | 4686 | 592.7 | 1620.48 | 2777403 | 7399 | 350.75 | 987.77 | 2595177 |
| Total | 20557 | 370.61 | 991.14 | 7618690 | 53403 | 168.17 | 531.35 | 8980612 |
Overall, the number of inserted and deleted bases is on par with what one would expect from a diploid sample ( e.g. http://genome.cshlp.org/content/early/2016/11/25/gr.214007.116.full.pdf+html), though a little high.
The count of the deletion variants is very high, driven by the STR and Complex callsets (a complex call is complex sequence context, e.g. unmasked bases, rather than a complex set of operations). The causes for this could be the use of p-reads as opposed to some more sophisticated model - larger events will be broken up into multiple smaller events in more noisy alignments, or due to a context dependent deletion error mode such as the loss of poly-a tracts.
Clive, if you care, I'll send the list of deletions that overlap with exons. The count is high, but it's driven by small events that are likely false positives. Not to worry though, nothing pops out.
The count of the deletion variants is very high, driven by the STR and Complex callsets (a complex call is complex sequence context, e.g. unmasked bases, rather than a complex set of operations). The causes for this could be the use of p-reads as opposed to some more sophisticated model - larger events will be broken up into multiple smaller events in more noisy alignments, or due to a context dependent deletion error mode such as the loss of poly-a tracts.
Clive, if you care, I'll send the list of deletions that overlap with exons. The count is high, but it's driven by small events that are likely false positives. Not to worry though, nothing pops out.
No comments:
Post a Comment