Friday, June 9, 2017
NYT parliamentary vote palette
Always on the hunt for a good 8 class palette, NYT has one for today's graphic of the parliamentary vote:
#FAA651 #FFEF4C #3989CB #D65454 #960630 #93C44C #6A996A #999999
Friday, January 6, 2017
SV Calling in the cliveome
Well, the CliveOme is out, and open for analysis. I took a few steps to do SV calling, and the results are presented below. Note this is very preliminary; a rough pass takes an afternoon, and nailing it all down months. In particular the counts for deletion are very high, and you see an imbalanced mobile element insertion and deletion rate (the rate should be the same).
1. Data.
The data I used was 53 fastq files taking up about 300G of space. I didn't plan on doing anything with the raw data.
2. Mapping:
I used my branch of blasr (https://github.com/mchaisso/blasr/commit/c950018b65728cf9a0806381f1e69fba6ea40b08)
to map to build 38, using a precomputed index
blasr input.fofn GRCh38.fasta -maxMatch 25 -advanceExactMatches 5 -sam -nproc 16 -minMapQV 30 -out /dev/stdout | samtools view -bS - | samtools sort -T /tmp/clive -o clive.bam
This took < 1 day using 16 cores and about 30G of memory.
3. Basic sequencing stats. The dataset had about 30X euchromatic coverage, with about 0.5Gbase unmapped. The data release quotes 50X, so there is a chance I didn't grab all the files.

5. Calling structural variation.
The current pipeline I use for calling SV on a diploid sample is: 1. phase SNVs. 2. Partition SMS reads counting overlap of phased SNVs, and 3. Calling SV on each set of partitioned reads separately. I didn't try taking the time to do the phasing because the SNV strand bias would cause a bunch of artifacts to appear in phase. The code to separate strand bias from real SNVs represented on both strands is running now, but it's /very/ slow and probably won't finish until long past my attention has turned back to my normal research.
I modified my pipeline to do local assembly in 60kbp tiling windows using Canu V1.4. Instead of mapping the local assemblies, the 10 longest p-reads from each local assembly were mapped, this helps fix the problem of detecting heterozygous variants. The p-reads had an average accurcy of 96.9%, making them unsuitable for indel calling and OK for calling SV. The p-reads were mapped back to hg38, and a set of unmerged SVs were output simply by parsing the cigars strings. To merge, SVs were split according to insertion and deletion, and then a graph was made with each SV as a node, and an edge connecting any two SVs if they had 50% reciprocal overlap. The largest SV from any connected component with at least two nodes was output.
3. Basic sequencing stats. The dataset had about 30X euchromatic coverage, with about 0.5Gbase unmapped. The data release quotes 50X, so there is a chance I didn't grab all the files.

While there are a few very long (> 100kbp) sequences here, it only accounts for 0.2% of the total coverage, and 8% of the total coverage allowing reads over 50kbp. This implies we can't start assuming de novo assembly with 100kbp reads yet.
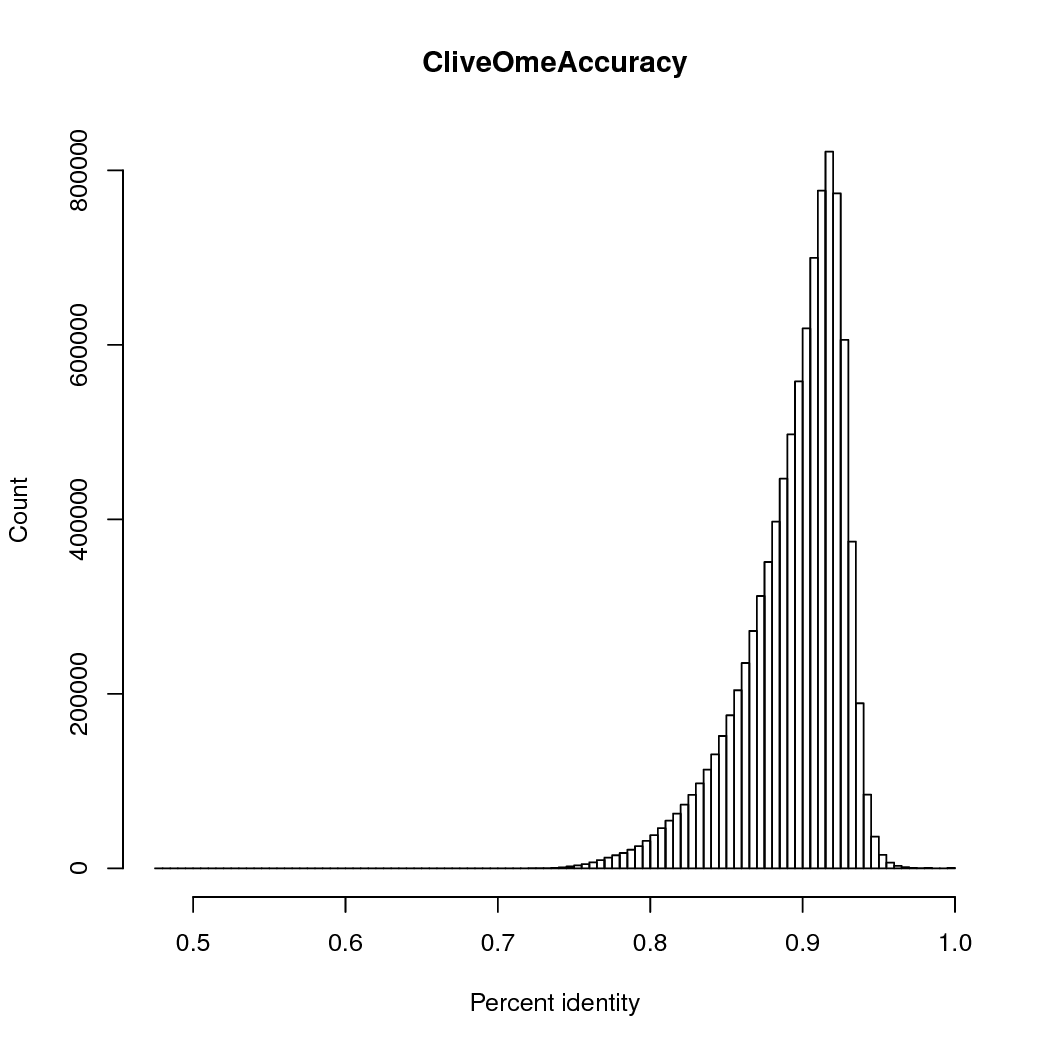
 The accuracy was 89%, counting # matches/ (#matches + # insertions + # deletions + # mismatches), so multi-base insertions or deletions are counted as single events.
The accuracy was 89%, counting # matches/ (#matches + # insertions + # deletions + # mismatches), so multi-base insertions or deletions are counted as single events.
 And like all SMS, there isn't a length/accuracy bias.
And like all SMS, there isn't a length/accuracy bias.
4. Sequencing error modes.
I'm a noob on nanopore sequencing, so much of what I may mention about error modes is probably well known. Generic regions look fine, but have a high SNV rate.

Zooming in on a few of these there was a pretty high strand bias for SNVs. Another way to look at this is, for each SNV, look at the ratio of the min(# forward, #reverse)/(#forward+#reverse) for all reads supporting an SNV, for the two haplotypes.

Clive mentioned on Twitter this could be due to modified bases and their basecaller not supporting native DNA yet, so I won't look any more to figure out what's happening.
Here is the strand ratio for the pacbio SNVs on chromosome 1 for a different individual:


4. Sequencing error modes.
I'm a noob on nanopore sequencing, so much of what I may mention about error modes is probably well known. Generic regions look fine, but have a high SNV rate.

Zooming in on a few of these there was a pretty high strand bias for SNVs. Another way to look at this is, for each SNV, look at the ratio of the min(# forward, #reverse)/(#forward+#reverse) for all reads supporting an SNV, for the two haplotypes.

Here is the strand ratio for the pacbio SNVs on chromosome 1 for a different individual:

Also, effectively all poly-A/polyT sequences I saw were deleted:


5. Calling structural variation.
The current pipeline I use for calling SV on a diploid sample is: 1. phase SNVs. 2. Partition SMS reads counting overlap of phased SNVs, and 3. Calling SV on each set of partitioned reads separately. I didn't try taking the time to do the phasing because the SNV strand bias would cause a bunch of artifacts to appear in phase. The code to separate strand bias from real SNVs represented on both strands is running now, but it's /very/ slow and probably won't finish until long past my attention has turned back to my normal research.
I modified my pipeline to do local assembly in 60kbp tiling windows using Canu V1.4. Instead of mapping the local assemblies, the 10 longest p-reads from each local assembly were mapped, this helps fix the problem of detecting heterozygous variants. The p-reads had an average accurcy of 96.9%, making them unsuitable for indel calling and OK for calling SV. The p-reads were mapped back to hg38, and a set of unmerged SVs were output simply by parsing the cigars strings. To merge, SVs were split according to insertion and deletion, and then a graph was made with each SV as a node, and an edge connecting any two SVs if they had 50% reciprocal overlap. The largest SV from any connected component with at least two nodes was output.
| Insertion | Deletion | |||||||
| Repeat type | Count | Mean | s.d. | Total | Count | Mean | s.d. | Total |
| Complex | 4968 | 118.91 | 191.23 | 590753 | 18616 | 83.75 | 121.19 | 1559030 |
| AluY | 1319 | 263.54 | 47.94 | 347611 | 1857 | 220.39 | 119.8 | 409259 |
| AluS | 183 | 176.63 | 79.68 | 32323 | 1589 | 95.21 | 57.34 | 151293 |
| L1Hs | 171 | 1971.84 | 2145.28 | 337185 | 259 | 1597.51 | 2240.74 | 413754 |
| L1 | 192 | 765.58 | 1695.6 | 146992 | 1734 | 148.16 | 393.03 | 256910 |
| L1P | 427 | 553.71 | 931.83 | 236436 | 2109 | 172 | 381.9 | 362747 |
| SVA | 256 | 709.81 | 841.54 | 181711 | 430 | 452.19 | 798.13 | 194442 |
| HERV | 44 | 288.02 | 422.82 | 12673 | 274 | 143.41 | 355.73 | 39295 |
| Alu-mosaic | 254 | 358.31 | 256.56 | 91010 | 589 | 166.86 | 195.64 | 98278 |
| STR | 4690 | 244.82 | 328.21 | 1148188 | 13312 | 84.65 | 65.21 | 1126812 |
| Satellite | 2583 | 297.44 | 482.81 | 768280 | 2645 | 221.41 | 460.67 | 585625 |
| Singletons | 308 | 208.39 | 367.86 | 64184 | 1898 | 103.76 | 140.33 | 196930 |
| Tandem_Repeats | 4686 | 592.7 | 1620.48 | 2777403 | 7399 | 350.75 | 987.77 | 2595177 |
| Total | 20557 | 370.61 | 991.14 | 7618690 | 53403 | 168.17 | 531.35 | 8980612 |
Overall, the number of inserted and deleted bases is on par with what one would expect from a diploid sample ( e.g. http://genome.cshlp.org/content/early/2016/11/25/gr.214007.116.full.pdf+html), though a little high.
The count of the deletion variants is very high, driven by the STR and Complex callsets (a complex call is complex sequence context, e.g. unmasked bases, rather than a complex set of operations). The causes for this could be the use of p-reads as opposed to some more sophisticated model - larger events will be broken up into multiple smaller events in more noisy alignments, or due to a context dependent deletion error mode such as the loss of poly-a tracts.
Clive, if you care, I'll send the list of deletions that overlap with exons. The count is high, but it's driven by small events that are likely false positives. Not to worry though, nothing pops out.
The count of the deletion variants is very high, driven by the STR and Complex callsets (a complex call is complex sequence context, e.g. unmasked bases, rather than a complex set of operations). The causes for this could be the use of p-reads as opposed to some more sophisticated model - larger events will be broken up into multiple smaller events in more noisy alignments, or due to a context dependent deletion error mode such as the loss of poly-a tracts.
Clive, if you care, I'll send the list of deletions that overlap with exons. The count is high, but it's driven by small events that are likely false positives. Not to worry though, nothing pops out.
Thursday, October 1, 2015
What does SEQUEL mean for human genetics?

Two milestones happened this week in genomics: the 1000-genomes project wrapped up, and pacbio announced a new machine. The 1kg papers are setting the new minimum for high-impact “large” genomics studies: cohorts on the order of 1000 genomes (funded by a dedicated R01 or consortium). Any large scale switch from Illumina would have to be able to sequence this many samples.
Genomics studies using pacbio sequencing will likely target structural variation since Amplified and Cycled Sequencing (ACS*, Illumina for example) is just fine for SNVs. My current methods for structural detection need more than 10x coverage, though they were hastily written and I will try to make coverage requirements go down with better methods.
For the time being, for a 30x genome on sequel, many rough estimates are placing a pacbio based human genome between $10k-20k. Before you spend any time debating that, remember this is a moving target with many variables for actual cost at play. For example, our cost center charges around $800/SMRTCell, and this includes overhead of labor and cost amortization of the instrument. The lower instrument cost and fixed labor cost means a 3x increase in SMRTCell doesn't translate to a 3x increase in total run cost.
For disorders where there is missing heritability, we may see some <100 genome pilot studies launched - consider the Gilissen et al paper http://www.ncbi.nlm.nih.gov/pubmed/24896178 which demonstrated an increase in diagnostic yield moving from WES to WGS. Still, much of the variation detected in this paper is coding regions missed by exome captures but picked up in WGS. The 3 percent or so of genes missed by ACS reads isn't likely to explain the remainder of missing heritability, and so we'll be looking for new variation in noncoding sequences.
Large scale WES studies were pretty much guaranteed to have significant results -- either the causal mutations in Mendelian disorders or enough likely causal genes in simplex cohorts to improve our understanding of a disease. The noncoding space is a bit more unpredictable. There are plenty of examples of disrupted enhancers. The SHH mutation in polydactyly comes to mind, though this is a point mutation. If more examples of small SVs taking out functional regulatory elements are found, it can certainly help increase our understanding of regulatory architecture. Also, consider that the FTD-ALS locus was not detected until several years ago after a massive dedicated effort from multiple groups. The initial human SMS studies are finding lots of tandem repeat variation, perhaps there are other disorders lurking in our genomes?
The short answer is that an SMS based study of human disease is almost guaranteed. I apologize for the trite ending, but in the long run, the question will be what will human genetics mean for SEQUEL?
*This is my acronym for NGS/HTS/short-reads, and I'd love for it to catch on.
Wednesday, September 2, 2015
Same same, but different

Historically there has been a pretty deep chasm between the two, and for trainees about to finish their Ph.D. making this decision may seem like choosing between the red and the blue pill. There are plenty of differences but perhaps more similarities now than there used to be.
Science and innovation. A common characterization of industrial work is that it often involves perfecting a product, rather than innovating it. If this is more appealing to you than the work you did for your thesis, there are ample opportunities to do so in industry and that is probably the right choice for you.
If you are pursuing 'pure' science, then academia is the right path, and you too do not need to continue reading. For the rest of us, much of bioinformatics research is applied science, and the boundaries between what you do in research and what you could do in industry are somewhat blurred. Case in point the two competing assemblers for PacBio data are HGAP/Falcon (industry) and MHAP (academia), but both have authors from each side on publications.
Businesses may gain greater acceptance in the scientific community if their research is accepted as equal by scientific peers than if their sole communiques are through marketing. In this way, it is possible to do innovative research in industry, but the main difference to get used to from your training is it must be ultimately directed at increasing revenue at the company, and you have less freedom in deciding the direction of your work.
The direct parallel in academia is that your research needs to feed into your next grant application. True, you should maintain a coherent research thread that supports likelihood of being able to carry out your aims in your grant application. Same same, but different. Doing new research in both academia and industry involves convincing some sort of board your work is worth funding, but academia is built upon new research, and it is a bit more difficult to take on new directions in industry.
Job stability. NIH funding has the stability of a roller coaster, but it is not safe to assume your position in industry is as solid as bedrock either. In both, you need to make sure your skills and knowledge are state of the art. In industry it seems there is a trade off between risk and innovation, where companies are more likely to cut experimental branches of their company that may have been more intellectually stimulating than their core counter parts. If you are adamant about not going into academia, the environment to join a startup is a good alternative right now. You will have to have laser focus on getting the startup running, but it is at least as challenging as your thesis work, and the lack of job stability from a startup is compensated by the number of opportunities to move to if the first you start or join does not work out.
Organization. Academic research by nature tends to be a bit more... disorganized. Maybe compare the cathedral and the bazaar analogy of open source development. If you are going to go into industry, be prepared to face the Gantt chart and the program manager. This is not for the scientific faint of heart. Looking at a monitor showing an organized timeline of conservatively chosen milestones that dictate the next 3-6 months of your work, some may see relief, and others a straight jacket.
Work-life balance. With some exaggeration, one can say the work-life balance in academia is perfectly balanced, as long as your work and life are the same. But in seriousness, it is much easier to put in a 9-5 day in industry than academia and I tend to see people who have a non-scientific passion more satisfied in industry. As with everything I've stated before, there are plenty of exceptions, but I often saw in the bay area a trade-off between work that was not very engaging, but allowed time for other activities, and all-consuming positions such as what you often see in faculty. Pavel Pevzner addressed this in a great commencement address at SFU in 2011: as long as you're following your passion, you don't notice you're working at all! It is not a completely dire situation, just the balance in this part is tipped a bit towards industry.
Wednesday, September 11, 2013
Converting bas.h5 to fasta with pls2fasta.
I would strongly recommend taking the time to install pbcore/pbh5toos in order to work with bas/bax h5 files - these are kept up to date by the software group at pacbio.
If you're having difficulty getting this set up on your system, it is possible to use a binary compiled file pls2fasta to convert bas.h5 files into fasta and fastq format. This is part of the blasr source you can download from github. After compiling there will be an executable $(source_dir)/pbihdfutils/bin/pls2fasta
Typical usage is as follows:
Typical usage is as follows:
>pls2fasta in.bas.h5 out.fasta -trimByRegionThe flag "trimByRegion" is necessary to only include the high quality regions of reads in "out.fasta". The toher regions are not just low quality, but pure noise caused by signal recorded before sequencing began, or recorded after the true sequence ended. The key is that you don't gain any information by leaving in any of the low quality portions. To produce fastq output, use:
>pls2fasta in.bas.h5 out.fastq -trimByRegion -fastqI do not think there is software out there that handles pacbio reads that makes appropriate use of the quality values other than the Quiver method released by PacBio. It is possible to make blasr use quality values to form pairwise alignments, but by default this is turned off because the adjacent insertion/deletion columns that are supported by quality value aware alignment often cause problems to naive consensus calling methods.
Subscribe to:
Posts (Atom)